| Citation: |
Minyang Dai, Yan Zhang, Wenpeng Ni, Shiguo Zhang. A perspective on electrochemical conversion of CO2 to multicarbon chemicals in ionic liquids-based electrolytes[J]. Energy Lab, 2023, 1(3): 230006. doi: 10.54227/elab.20230006

|
A perspective on electrochemical conversion of CO2 to multicarbon chemicals in ionic liquids-based electrolytes
-
Abstract
Carbon dioxide electroreduction (CRR) is a promising technology for both intermittent energy storage and emissions mitigation, but it faces challenges such as relatively high overpotential and poor selectivity. Introducing ionic liquids (ILs) into the CRR system has shown impressive activity for CO production, even in electrocatalysts that are primarily active for hydrogen evolution in aqueous electrolytes. However, converting CO2 to high-value C2+ chemicals in IL electrolytes suffers from limitations in *CO coverage, proton accessibility, and specific stabilization effects on *COOH. In this perspective, we emphasize the modification of the steady-state adsorption of *CO and other intermediates to enhance the CO2-to-C2+ conversion. More efforts need to be devoted to electrolyte modulation, involving the functional ILs design, the proton sources, and inorganic additive screening. It is also necessary to design effective electrocatalysts via developing a descriptor for C2+ selectivity, exploring the dynamic evolution of catalyst upon exposure to ILs, and constructing novel catalyst/ILs hybrids. Furthermore, developing a molecular understanding of the electrode/ILs interface and the bulk phase of IL-containing electrolytes could provide guildelines for designing an efficient electrochemical system for C2+ generation.
-
Keywords:
- ionic liquids /
- carbon dioxide /
- electrochemical reduction /
- multicarbon chemicals
-
-
Author Biographies
 Minyang Dai is pursuing her Ph.D degree under the direction of Prof. Shiguo Zhang at the College of Materials Science and Engineering, Hunan University. Her research focuses on the design of carbon materials and the application of ionic liquids for carbon dioxide electrochemical reduction.
Minyang Dai is pursuing her Ph.D degree under the direction of Prof. Shiguo Zhang at the College of Materials Science and Engineering, Hunan University. Her research focuses on the design of carbon materials and the application of ionic liquids for carbon dioxide electrochemical reduction.  Wenpeng Ni is currently a lecturer at the College of Materials Science and Engineering, Hunan University. He finished a successive postgraduate and doctoral program in 2018, and received his Ph.D degree from Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences. His research interests mainly include the construction of high-efficient electrocatalysis system for carbon dioxide reduction and organic small molecule conversion via interfacial molecular design.
Wenpeng Ni is currently a lecturer at the College of Materials Science and Engineering, Hunan University. He finished a successive postgraduate and doctoral program in 2018, and received his Ph.D degree from Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences. His research interests mainly include the construction of high-efficient electrocatalysis system for carbon dioxide reduction and organic small molecule conversion via interfacial molecular design.  Shiguo Zhang is currently a professor at the College of Materials Science and Engineering, Hunan University. He received his Ph.D degree in 2011, supervised by Prof. Youquan Deng, from the Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences (CAS), and worked as an assistant research in the LICP from 2011 to 2012. In April 2012, he joined Prof. Masayoshi Watanabe’s group as a postdoctoral worker at Yokohama National University. His research interests include ionic liquids, carbon materials and electrochemical energy conversion and storage.
Shiguo Zhang is currently a professor at the College of Materials Science and Engineering, Hunan University. He received his Ph.D degree in 2011, supervised by Prof. Youquan Deng, from the Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences (CAS), and worked as an assistant research in the LICP from 2011 to 2012. In April 2012, he joined Prof. Masayoshi Watanabe’s group as a postdoctoral worker at Yokohama National University. His research interests include ionic liquids, carbon materials and electrochemical energy conversion and storage. -
References
1. G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen, L. Dai, Chem. Soc. Rev., 2021, 50, 4993 2. M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton, E. H. Sargent, Adv. Mater., 2019, 31, e1807166 3. A. R. Woldu, Z. Huang, P. Zhao, L. Hu, D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 4. Y. Xue, Y. Guo, H. Cui, Z. Zhou, Small Methods, 2021, 5, 2100736 5. B. Deng, M. Huang, X. Zhao, S. Mou, F. Dong, ACS Catal., 2022, 12, 331 6. M. Moura de Salles Pupo, R. Kortlever, ChemPhysChem, 2019, 20, 2926 7. D. Gao, R. M. Arán-Ais, H. S. Jeon, B. Roldan Cuenya, Nat. Catal., 2019, 2, 198 8. Y. Huang, C. W. Ong, B. S. Yeo, ChemSusChem, 2018, 11, 3299 9. D. Gao, I. T. McCrum, S. Deo, Y.-W. Choi, F. Scholten, W. Wan, J. G. Chen, M. J. Janik, B. Roldan Cuenya, ACS Catal., 2018, 8, 10012 10. H.-K. Lim, Y. Kwon, H. S. Kim, J. Jeon, Y.-H. Kim, J.-A. Lim, B.-S. Kim, J. Choi, H. Kim, ACS Catal., 2018, 8, 2420 11. S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell, K. Chan, Energy Environ. Sci., 2019, 12, 3001 12. M. Konig, J. Vaes, E. Klemm, D. Pant, iScience, 2019, 19, 135 13. Z. Lei, B. Chen, Y.-M. Koo, D. R. Macfarlane, Chem. Rev., 2017, 117, 6633 14. G.-R. Zhang, B. J. M. Etzold, J. Energy Chem., 2016, 25, 199 15. G. Iijima, T. Kitagawa, A. Katayama, T. Inomata, H. Yamaguchi, K. Suzuki, K. Hirata, Y. Hijikata, M. Ito, H. Masuda, ACS Catal., 2018, 8, 1990 16. Y. Sha, J. Zhang, X. Cheng, M. Xu, Z. Su, Y. Wang, J. Hu, B. Han, L. Zheng, Angew. Chem. Int. Ed., 2022, 61, e202200039 17. Y. Wu, C. Chen, X. Yan, X. Sun, Q. Zhu, P. Li, Y. Li, S. Liu, J. Ma, Y. Huang, B. Han, Angew. Chem. Int. Ed., 2021, 60, 20803 18. P. Li, J. Bi, J. Liu, Q. Zhu, C. Chen, X. Sun, J. Zhang, B. Han, Nat. Commun., 2022, 13, 1965 19. J. Medina-Ramos, R. C. Pupillo, T. P. Keane, J. L. DiMeglio, J. Rosenthal, J. Am. Chem. Soc., 2015, 137, 5021 20. M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Král, J. Abiade, A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470 21. B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin, A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819 22. X. Sun, L. Lu, Q. Zhu, C. Wu, D. Yang, C. Chen, B. Han, Angew. Chem. Int. Ed., 2018, 57, 2427 23. J. L. DiMeglio, J. Rosenthal, J. Am. Chem. Soc., 2013, 135, 8798 24. Y. Hori, I. Takahashi, O. Koga, N. Hoshi, J. Phys. Chem. B, 2002, 106, 15 25. J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu, Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 26. Y. Cui, B. He, X. Liu, J. Sun, Ind. Eng. Chem. Res., 2020, 59, 20235 27. A. V. Rudnev, Y. C. Fu, I. Gjuroski, F. Stricker, J. Furrer, N. Kovacs, S. Vesztergom, P. Broekmann, ChemPhysChem, 2017, 18, 3153 28. B. A. Rosen, J. L. Haan, P. Mukherjee, B. Braunschweig, W. Zhu, A. Salehi-Khojin, D. D. Dlott, R. I. Masel, J. Phys. Chem. C, 2012, 116, 15307 29. Y. Wang, M. Hatakeyama, K. Ogata, M. Wakabayashi, F. Jin, S. Nakamura, Phys. Chem. Chem. Phys., 2015, 17, 23521 30. W. Ge, Y. Chen, Y. Fan, Y. Zhu, H. Liu, L. Song, Z. Liu, C. Lian, H. Jiang, C. Li, J. Am. Chem. Soc., 2022, 144, 6613 31. J. T. Feaster, A. L. Jongerius, X. Liu, M. Urushihara, S. A. Nitopi, C. Hahn, K. Chan, J. K. Norskov, T. F. Jaramillo, Langmuir, 2017, 33, 9464 32. D. V. Vasilyev, S. Shyshkanov, E. Shirzadi, S. A. Katsyuba, M. K. Nazeeruddin, P. J. Dyson, ACS Appl. Energy Mater., 2020, 3, 4690 33. D. Vasilyev, E. Shirzadi, A. V. Rudnev, P. Broekmann, P. J. Dyson, ACS Appl. Energy Mater., 2018, 1, 5124 34. E. E. L. Tanner, C. Batchelor-McAuley, R. G. Compton, J. Phys. Chem. C, 2016, 120, 26442 35. S.-F. Zhao, M. Horne, A. M. Bond, J. Zhang, J. Phys. Chem. C, 2016, 120, 23989 36. J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan, A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277 37. L. Gao, L. Qin, H. Wu, X. Li, K. Qi, Q. Yi, J. Zhang, L. Shi, Fuel, 2022, 322, 124103 38. D. J. Tao, F. F. Chen, Z. Q. Tian, K. Huang, S. M. Mahurin, D. E. Jiang, S. Dai, Angew. Chem. Int. Ed., 2017, 56, 6843 39. A. Marjolin, J. A. Keith, ACS Catal., 2015, 5, 1123 40. B. A. Rosen, W. Zhu, G. Kaul, A. Salehi-Khojin, R. I. Masel, J. Electrochem. Soc., 2013, 160, H138 41. H. Wu, J. Song, C. Xie, Y. Hu, B. Han, Green Chem., 2018, 20, 1765 42. A. Atifi, D. W. Boyce, J. L. DiMeglio, J. Rosenthal, ACS Catal., 2018, 8, 2857 43. A. T. Chu, Y. Surendranath, J. Am. Chem. Soc., 2022, 144, 5359 44. Z. Han, D. Han, Z. Chen, J. Gao, G. Jiang, X. Wang, S. Lyu, Y. Guo, C. Geng, L. Yin, Z. Weng, Q. H. Yang, Nat. Commun., 2022, 13, 3158 45. M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager, 3rd, A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006 46. C. M. Gunathunge, V. J. Ovalle, M. M. Waegele, Phys. Chem. Chem. Phys., 2017, 19, 30166 47. D. Gao, F. Scholten, B. Roldan Cuenya, ACS Catal., 2017, 7, 5112 48. T. N. Huan, P. Simon, G. Rousse, I. Genois, V. Artero, M. Fontecave, Chem. Sci., 2017, 8, 742 49. Q. Zhu, D. Yang, H. Liu, X. Sun, C. Chen, J. Bi, J. Liu, H. Wu, B. Han, Angew. Chem. Int. Ed., 2020, 8896 50. X. Kang, X. Sun, Q. Zhu, X. Ma, H. Liu, J. Ma, Q. Qian, B. Han, Green Chem., 2016, 18, 1869 51. D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu, B. Han, Nat. Commun., 2019, 10, 677 52. L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang, B. Han, Angew. Chem. Int. Ed., 2018, 57, 14149 53. X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, J. Ma, Z. Zhang, G. Yang, B. Han, Green Chem., 2017, 19, 2086 54. A. A. Peterson, J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251 55. J. Hussain, H. Jónsson, E. Skúlason, ACS Catal., 2018, 8, 5240 56. N. Zhang, B. Yang, K. Liu, H. Li, G. Chen, X. Qiu, W. Li, J. Hu, J. Fu, Y. Jiang, M. Liu, J. Ye, Small Methods, 2021, 5, 2100987 57. A. Bagger, W. Ju, A. S. Varela, P. Strasser, J. Rossmeisl, Chemphyschem, 2017, 18, 3266 58. Z. Zhao, Z. Chen, X. Zhang, G. Lu, J. Phys. Chem. C, 2016, 120, 28125 59. M. Yang, Z. Wang, D. Jiao, Y. Tian, Y. Shang, L. Yin, Q. Cai, J. Zhao, J. Energy Chem., 2022, 69, 456 60. P. Chen, P. Zhang, X. Kang, L. Zheng, G. Mo, R. Wu, J. Tai, B. Han, J. Am. Chem. Soc., 2022, 144, 14769 61. X.-Q. Li, G.-Y. Duan, J.-W. Chen, L.-J. Han, S.-J. Zhang, B.-H. Xu, Appl. Catal. B Environ., 2021, 297, 120471 62. H. K. Lim, H. Kim, Molecules, 2017, 22, 536 63. B. Braunschweig, P. Mukherjee, J. L. Haan, D. D. Dlott, J. Electroanal. Chem., 2017, 800, 144 -
Rights and permissions
This is an open access article under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Figures(7)
Tables(1)
Information
Article Metrics

Export File
Citation
Minyang Dai, Yan Zhang, Wenpeng Ni, Shiguo Zhang. A perspective on electrochemical conversion of CO2 to multicarbon chemicals in ionic liquids-based electrolytes[J]. Energy Lab, 2023, 1(3): 230006. doi: 10.54227/elab.20230006
Format
Content
 DownLoad:
DownLoad:
-
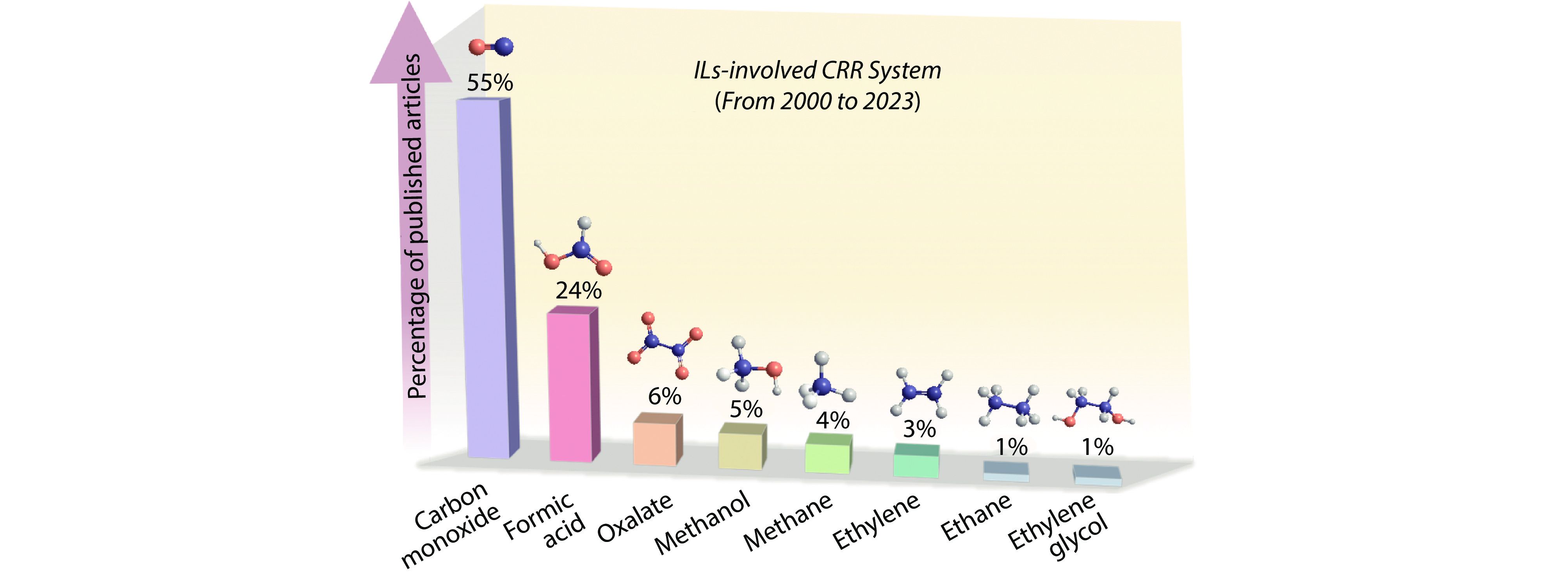
Figure 1.
Percentage of published articles on various CRR products in IL-containing systems.
-
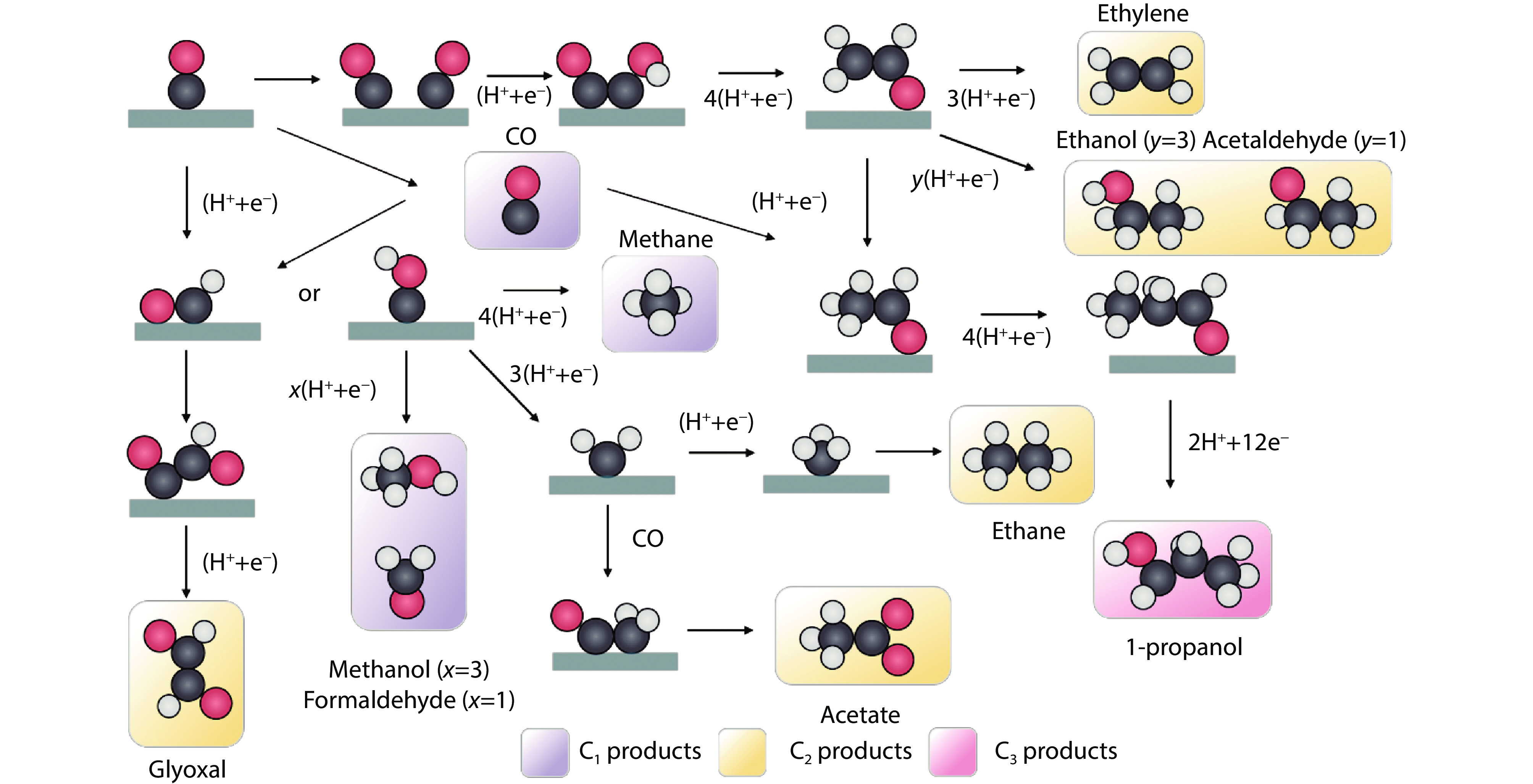
Figure 2.
Reaction pathways for CRR towards different products. (Black, C; red, O; grey, H; green, catalyst).
-
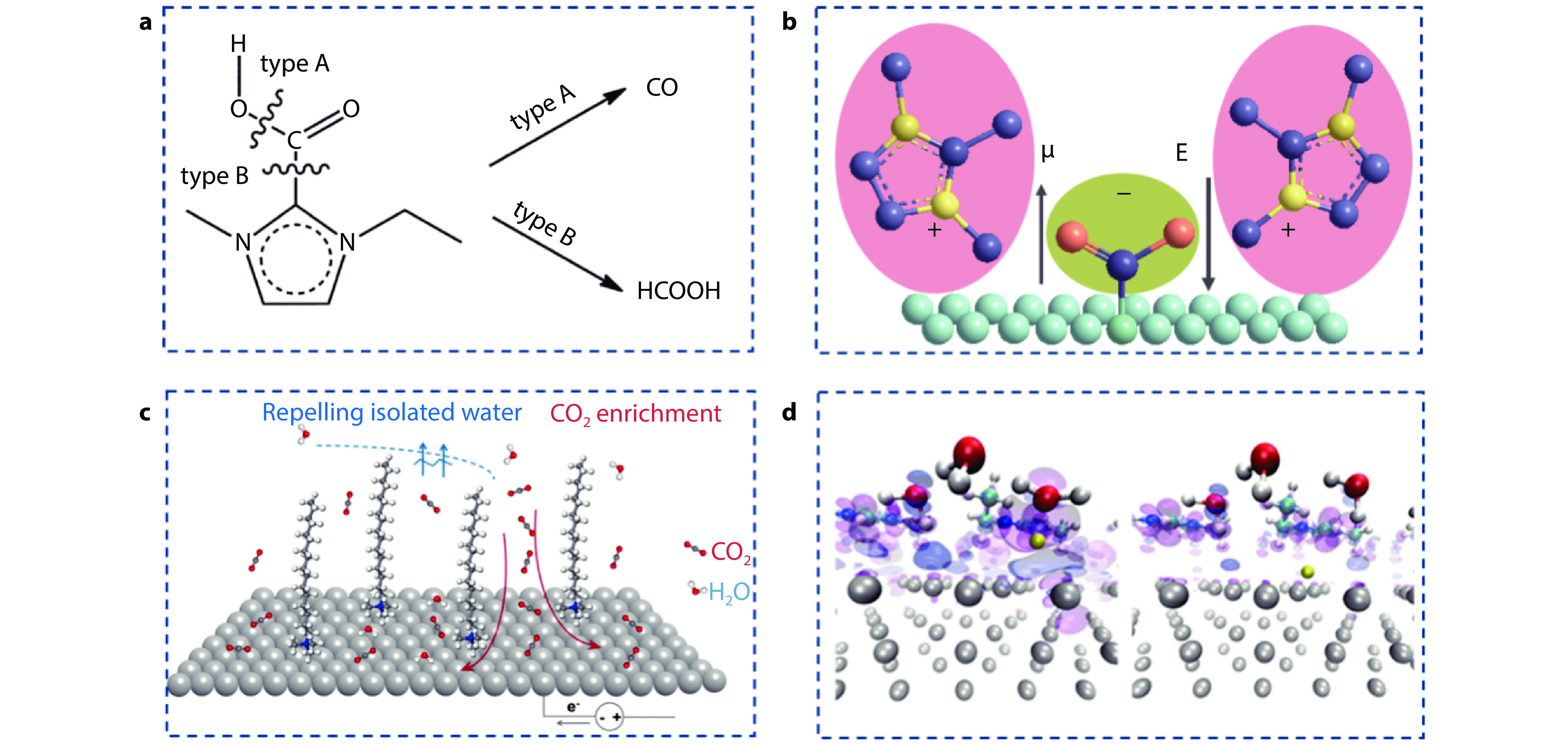
Figure 3.
a Covalent interaction between [EMIm] and intermediate *COOH.[29] Copyright 2015, RSC Publisher. b Stabilization of CO2•− by field-dipole interaction. c Configuration of CTAB at the electrified electrode−electrolyte interface.[30] Copyright 2022, American Chemical Society. d Charge density isosurfaces for transition, and final states of the Volmer reaction on bare [EMIM]+ -covered (bottom row) Ag(111). Magenta part represents an isosurface with a ρ value of 0.001 e b−3 and blue corresponds to a ρ value of -0.001 e b−3. The yellow one is the H atom being transferred along the reaction path.[31] Copyright 2017, American Chemical Society.
-
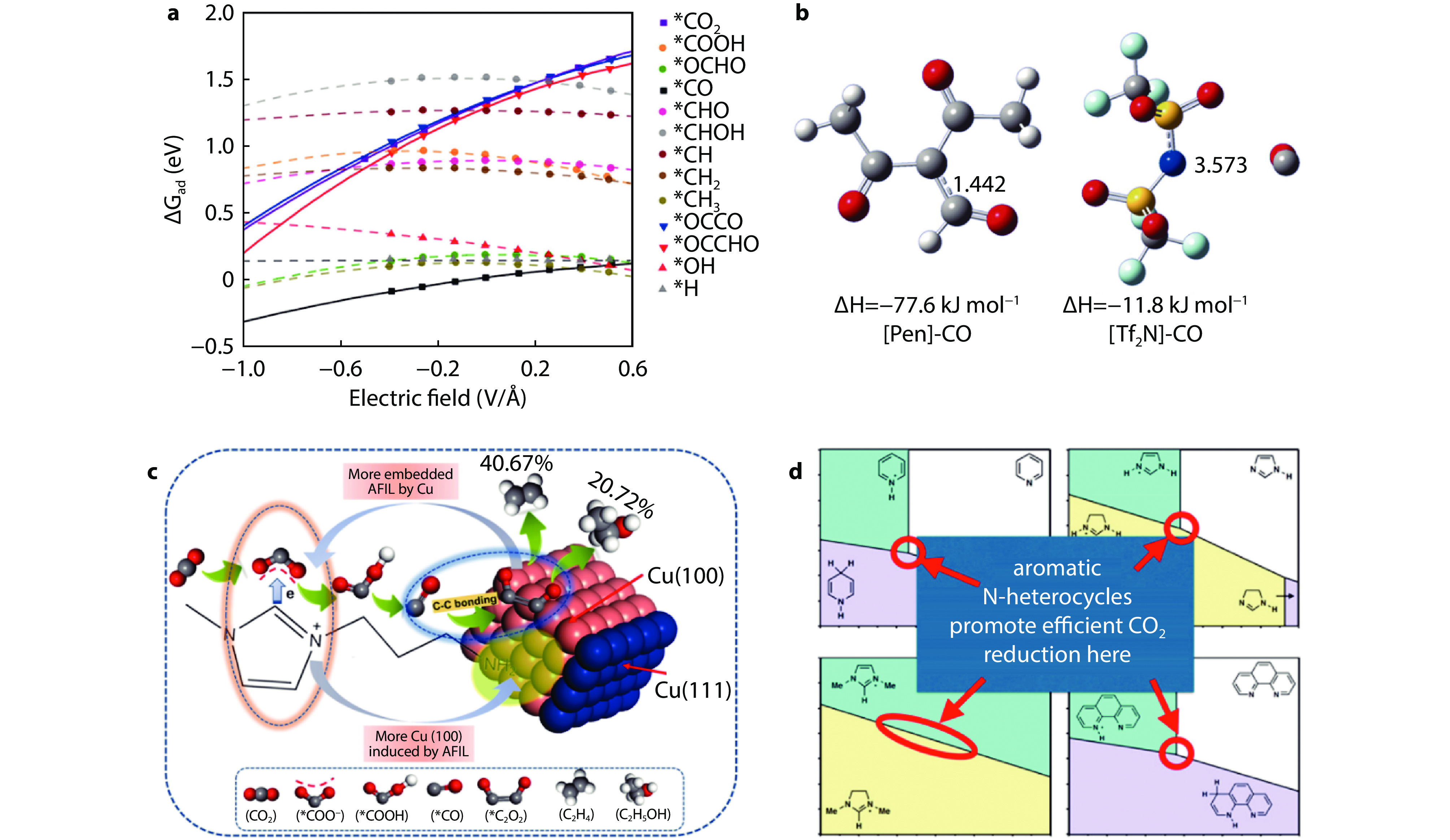
Figure 4.
a Field effect on various intermediates for CRR.[36] Copyright 2017, American Chemical Society. b Optimized structures of [Pen]-CO and [Tf2N]-CO.[38] Copyright 2017, Wiley-VCH. c Scheme for the synergistic effect between Cu and AFIL on the possible pathway during the CRR to C2.[37] Copyright 2022, Elsevier Ltd. d Molecular Pourbaix diagrams for aromatic N-heterocycles.[39] Copyright 2015, American Chemical Society.
-
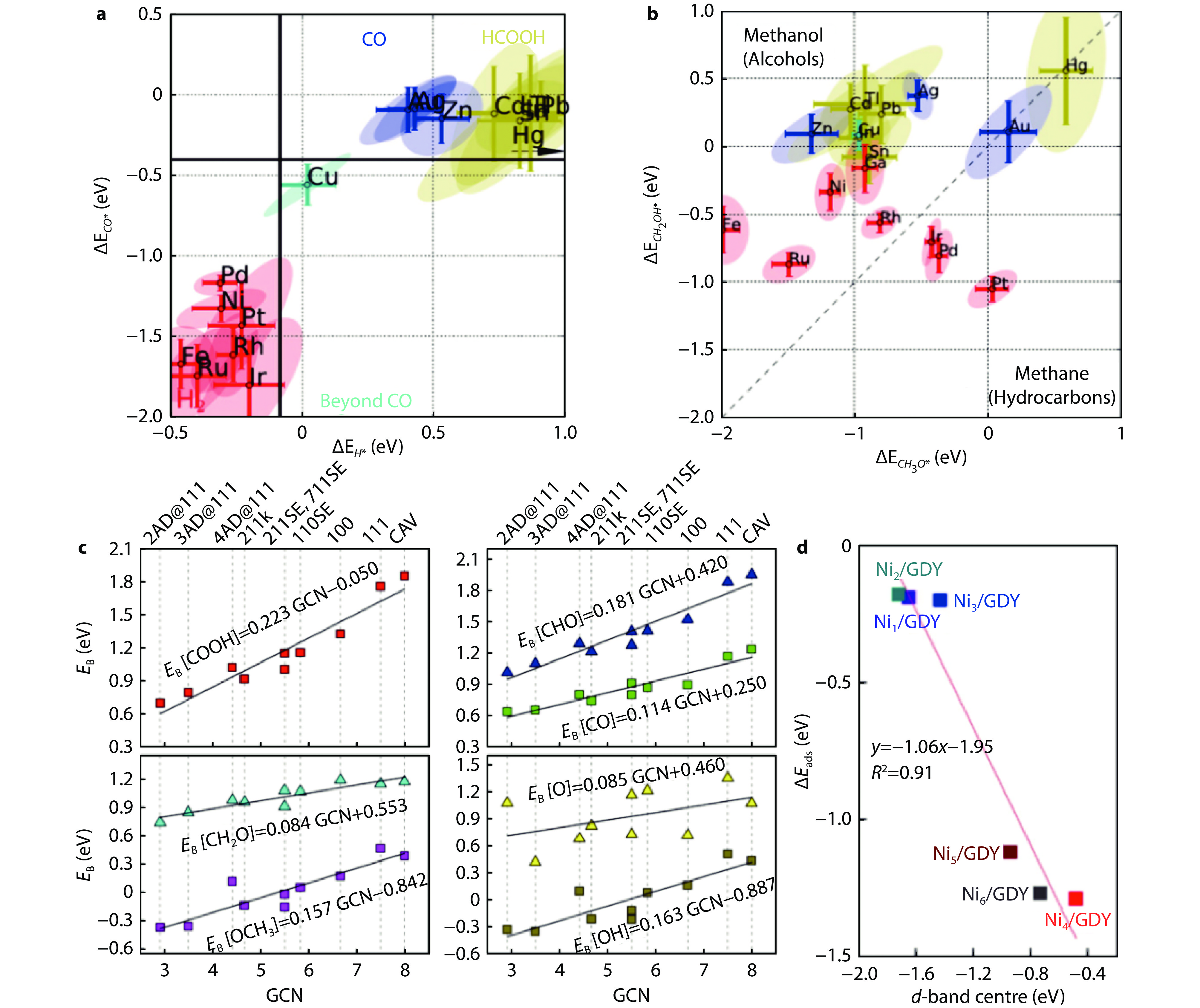
Figure 5.
Binding energies of a *CO and *H and b *CH2OH and *CH3O of various metals.[57] Copyright 2017, Wiley-VCH. c The linear scaling relations between generalized coordination number (GCN) and binding energy of intermediates.[58] Copyright 2016, American Chemical Society. d The variation of CO2 adsorption energy with d-band center.[59] Copyright 2022, Elsevier B.V. and Science Press.
-
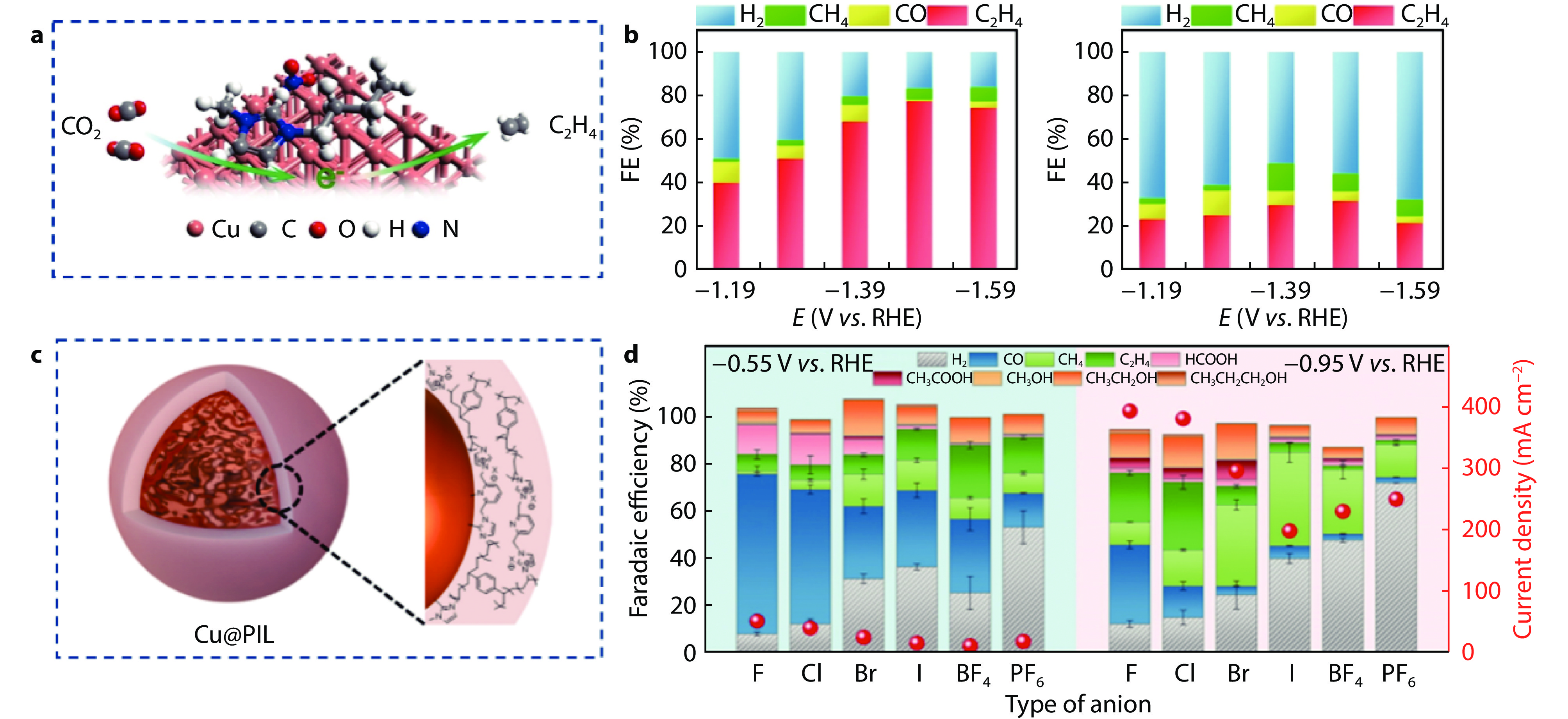
Figure 6.
a The model and b Faradaic efficiency of Cu and BMMImNO3@Cu.[61] Copyright 2021, Elsevier B.V. c The model and d Faradaic efficiency of Cu@PIL-X-1.2 with different anions.[16] Copyright 2022, Elsevier Ltd.
