| Citation: |
Xiaoming Xu, Yuanming Zhang, Yong Chen, Changhao Liu, Yang Li, Zhonghua Li, Zhetong Yang, Zhaosheng Li, Zhigang Zou. Green organic conversion with H2O2: challenges and opportunities[J]. Energy Lab, 2024, 2(1): 230011. doi: 10.54227/elab.20230011

|
Green organic conversion with H2O2: challenges and opportunities
-
Abstract
Hydrogen peroxide (H2O2), as a green oxidant, plays an important role in organic conversion reactions, such as cyclohexanone ammoximation and olefin oxidation. However, the production of H2O2 relies on the anthraquinone process, which is costly, complex, and typically done on clustered production. Furthermore, H2O2 is prone to decomposition or the generation of ineffective byproducts and unfavorable reactive groups, leading to low efficiency and waste of resources. Achieving the widespread application of H2O2 in green organic conversion reactions requires efficient utilization and low-cost on-site production of H2O2. Effective activation of H2O2 is the key to realizing efficient utilization of H2O2, which has been widely recognized. In addition, some emerging methods of on-site production of H2O2 are convenient and low-cost. These methods may gradually overcome the shortcomings of traditional methods in the future. In this review, we introduce common organic conversion reactions with H2O2, summarize the challenges of H2O2 activation, and review the progress on electrochemical, photoelectrochemical or photochemical H2O2 production. We also discuss the vision of organic conversion reactions via in-situ-generated H2O2.
-
Keywords:
- organic conversion /
- H2O2 production /
- activation of H2O2 /
- in-situ-generated H2O2
-
-
Author Biographies
 Xiaoming Xu obtained his bachelor’s degree in Materials Science and Engineering from Zhengzhou University in 2019. Now, he is a Ph.D. student at Nanjing University under the supervision of Prof. Zhaosheng Li. His research interest mainly focuses on the study of H2O2 activation and utilization.
Xiaoming Xu obtained his bachelor’s degree in Materials Science and Engineering from Zhengzhou University in 2019. Now, he is a Ph.D. student at Nanjing University under the supervision of Prof. Zhaosheng Li. His research interest mainly focuses on the study of H2O2 activation and utilization.  Zhaosheng Li received his Ph.D. degree in Condensed Matter Physics from the Institute of Solid State Physics, Chinese Academy of Sciences, in 2003. After a two-year postdoctoral fellowship at Nanjing University, he became a Lecturer at this university. In 2006, he was promoted to Associate Professor. Since 2011, he has become a full Professor of Materials Science and Engineering at the College of Engineering and Applied Sciences, Nanjing University. His current research interest includes photochemistry and photocatalysis.
Zhaosheng Li received his Ph.D. degree in Condensed Matter Physics from the Institute of Solid State Physics, Chinese Academy of Sciences, in 2003. After a two-year postdoctoral fellowship at Nanjing University, he became a Lecturer at this university. In 2006, he was promoted to Associate Professor. Since 2011, he has become a full Professor of Materials Science and Engineering at the College of Engineering and Applied Sciences, Nanjing University. His current research interest includes photochemistry and photocatalysis. -
References
1. T. Punniyamurthy, S. Velusamy, J. Iqbal, Chem. Rev., 2005, 105, 2329 2. J. Schranck, A. Tlili, M. Beller, Angew. Chem. Int. Ed., 2013, 52, 7642 3. Y. Shi, Y. Xia, Wen Langyou, L. Gao, G. Xu, B. Zong, Chem. Ind. Eng. Prog., 2021, 40, 2048 4. A. Aquino, O. Korup, R. Horn, Ind. Eng. Chem. Res., 2023, 62, 3098 5. Y. Shi, Y. Xia, G. Xu, L. Wen, G. Gao, B. Zong, Chinese J. Chem. Eng., 2022, 41, 145 6. A. A. Ingle, S. Z. Ansari, D. Z. Shende, K. L. Wasewar, A. B. Pandit, Environ. Sci. Pollut. Res. Int., 2022, 29, 86468 7. C. P. Gordon, H. Engler, A. S. Tragl, M. Plodinec, T. Lunkenbein, A. Berkessel, J. H. Teles, A. N. Parvulescu, C. Coperet, Nature, 2020, 586, 708 8. R. Vithalani, D. S. Patel, C. K. Modi, V. Sharma, P. K. Jha, Appl. Organomet. Chem., 2020, 34, e5500 9. X. Xu, Y. Zhang, Y. Chen, C. Liu, W. Wang, J. Wang, H. Huang, J. Feng, Z. Li, Z. Zou, Proc. Natl. Acad. Sci., 2022, 119, e2205562119 10. S. C. Perry, D. Pangotra, L. Vieira, L. I. Csepei, V. Sieber, L. Wang, C. Ponce de León, F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442 11. C. Xia, J. Y. Kim, H. Wang, Nat. Catal., 2020, 3, 605 12. C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen, G. J. Hutchings, ACS Catal., 2021, 11, 2701 13. R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. W. Chen, J. Z. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto, G. J. Hutchings, Science, 2022, 376, 615 14. M. Taramasso, G. Perego, B. Notari, U. S. Patent: 4, 410, 501[P], 1983. 15. X. Liang, Z. Mi, Y. Wang, L. Wang, X. Zhang, W. Wu, E. Min, S. Fu, J. Chem. Technol. Biot., 2004, 79, 658 16. G. Blanco-Brieva, M. C. Capel-Sanchez, M. P. de Frutos, A. Padilla-Polo, J. M. Campos-Martin, J. L. G. Fierro, Ind. Eng. Chem. Res., 2008, 47, 8011 17. T. Cousin, G. Chatel, N. Kardos, B. Andrioletti, M. Draye, Catal. Sci. Technol., 2019, 9, 5256 18. K. Dong, Y. Wang, L. Zhang, X. Fan, Z. Li, D. Zhao, L. Yue, S. Sun, Y. Luo, Q. Liu, A. A. Alshehri, Q. Li, D. Ma, X. Sun, Green Chem., 2022, 24, 8264 19. S. Tian, C. Peng, J. Dong, Q. Xu, Z. Chen, D. Zhai, Y. Wang, L. Gu, P. Hu, H. Duan, D. Wang, Y. Li, ACS Catal., 2021, 11, 4946 20. X. Li, Q. Wang, J. Lyu, X. Li, ChemistrySelect, 2021, 6, 9735 21. Y. Wan, Q. Liang, Z. Li, S. Xu, X. Hu, Q. Liu, D. Lu, J. Mol. Catal. A Chem., 2015, 402, 29 22. S. Ito, Y. Kon, T. Nakashima, D. Hong, H. Konno, D. Ino, K. Sato, Molecules, 2019, 24, 2520 23. C. Che, W. Yip, W. Yu, Chem. Asian J., 2006, 1, 453 24. M. N. Timofeeva, O. A. Kholdeeva, S. H. Jhung, J. S. Chang, Appl. Catal. A, 2008, 345, 195 25. Y. Zhang, X. Dai, J. Wang, J. Liang, J. Rabeah, X. Tian, X. Yao, Y. Wang, S. Pang, ChemSusChem, 2023, 16, e202202104 26. Y. Zeng, T. Chen, X. Zhang, Y. Chen, H. Zhou, L. Yu, Appl. Organomet. Chem., 2022, 36, e6658 27. L. Balapoor, R. Bikas, M. Dargahi, Inorg. Chim. Acta, 2020, 510, 119734 28. Y. Kon, T. Nakashima, A. Yada, T. Fujitani, S. Y. Onozawa, S. Kobayashi, K. Sato, Org. Biomol. Chem., 2021, 19, 1115 29. J. Wang, H. Yu, Z. Wei, Q. Li, W. Xuan, Y. Wei, Research, 2020, 2020, 3875920 30. Y. Zhang, N. Zhang, T. Wang, H. Huang, Y. Chen, Z. Li, Z. Zou, Appl. Catal. B, 2019, 245, 410 31. N. V. Maksimchuk, V. Y. Evtushok, O. V. Zalomaeva, G. M. Maksimov, I. D. Ivanchikova, Y. A. Chesalov, I. V. Eltsov, P. A. Abramov, T. S. Glazneva, V. V. Yanshole, O. A. Kholdeeva, R. J. Errington, A. Solé-Daura, J. M. Poblet, J. J. Carbó, ACS Catal., 2021, 11, 10589 32. N. V. Maksimchuk, I. D. Ivanchikova, G. M. Maksimov, I. V. Eltsov, V. Y. Evtushok, O. A. Kholdeeva, D. Lebbie, R. J. Errington, A. Solé-Daura, J. M. Poblet, J. J. Carbó, ACS Catal., 2019, 9, 6262 33. J. Li, C. Xiao, K. Wang, Y. Li, G. Zhang, Environ. Sci. Technol., 2019, 53, 11023 34. H. Zheng, Y. Zeng, J. Chen, R. Lin, W. Zhuang, R. Cao, Z. J. Lin, Inorg. Chem., 2019, 58, 6983 35. C. Ling, X. Liu, H. Li, X. Wang, H. Gu, K. Wei, M. Li, Y. Shi, H. Ben, G. Zhan, C. Liang, W. Shen, Y. Li, J. Zhao, L. Zhang, Angew. Chem. Int. Ed., 2022, 61, e202200670 36. Z. Yang, J. Qian, A. Yu, B. Pan, Proc. Natl. Acad. Sci., 2019, 116, 6659 37. Q. Yan, C. Lian, K. Huang, L. Liang, H. Yu, P. Yin, J. Zhang, M. Xing, Angew. Chem. Int. Ed., 2021, 60, 17155 38. Y. Xue, Y. Wang, Z. Pan, K. Sayama, Angew. Chem. Int. Ed., 2021, 60, 10469 39. N. Karamoschos, D. Tasis, Energies, 2022, 15, 6202 40. H. Lu, X. Li, S. A. Monny, Z. Wang, L. Wang, Chinese J. Catal., 2022, 43, 1204 41. X. Shi, S. Back, T. M. Gill, S. Siahrostami, X. Zheng, Chem, 2021, 7, 38 42. S. Li, J. Ma, F. Xu, L. Wei, D. He, Chem. Eng. J., 2023, 452, 139371 43. E. Berl1, Trans. Electrochem. Soc., 1939, 76, 359 44. C. Xia, Y. Xia, P. Zhu, L. Fan, H. Wang, Science, 2019, 366, 226 45. K. Wu, D. Wang, X. Lu, X. Zhang, Z. Xie, Y. Liu, B.-J. Su, J.-M. Chen, D.-S. Su, W. Qi, S. Guo, Chem, 2020, 6, 1443 46. Y. Wen, T. Zhang, J. Wang, Z. Pan, T. Wang, H. Yamashita, X. Qian, Y. Zhao, Angew. Chem. Int. Ed., 2022, 61, e202205972 47. Y. Kondo, Y. Kuwahara, K. Mori, H. Yamashita, Chem, 2022, 8, 2924 48. Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu, Y. Zhu, Nat. Energy, 2023, 8, 361 49. H. Yang, C. Li, T. Liu, T. Fellowes, S. Y. Chong, L. Catalano, M. Bahri, W. Zhang, Y. Xu, L. Liu, W. Zhao, A. M. Gardner, R. Clowes, N. D. Browning, X. Li, A. J. Cowan, A. I. Cooper, Nat. Nanotechnol., 2023, 18, 307 50. W. Zhang, P. Zhang, Q. Yang, K. Li, Chinese J. Org. Chem., 2022, 42, 732 51. S. J. Freakley, S. Kochius, J. van Marwijk, C. Fenner, R. J. Lewis, K. Baldenius, S. S. Marais, D. J. Opperman, S. T. L. Harrison, M. Alcalde, M. S. Smit, G. J. Hutchings, Nat. Commun., 2019, 10, 4178 52. Z. Jiang, L. Wang, J. Lei, Y. Liu, J. Zhang, Appl. Catal. B, 2019, 241, 367 53. F. Li, Q. Shao, M. Hu, Y. Chen, X. Huang, ACS Catal., 2018, 8, 3418 54. A. Prieto, M. Palomino, U. Díaz, A. Corma, Appl. Catal. A, 2016, 523, 73 55. W. Lee, L. Lai, M. Cem Akatay, E. A. Stach, F. H. Ribeiro, W. N. Delgass, J. Catal., 2012, 296, 31 56. N. Callaghan and J. A. Sullivan, Appl. Catal. B, 2014, 146, 258 57. R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. Qin, T. E. Davies, D. J. Morgan, A. Stenner, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, L. Chen, Y. Yamamoto, G. J. Hutchings, ACS Catal., 2023, 13, 1934 58. R. J. Lewis, K. Ueura, Y. Fukuta, T. E. Davies, D. J. Morgan, C. B. Paris, J. Singleton, J. K. Edwards, S. J. Freakley, Y. Yamamoto, G. J. Hutchings, Green Chem., 2022, 24, 9496 59. J. Lyu, L. Niu, F. Shen, J. Wei, Y. Xiang, Z. Yu, G. Zhang, C. Ding, Y. Huang, X. Li, ACS Omega, 2020, 5, 16865 60. A. K. Sinha, S. Seelan, S. Tsubota, M. Haruta, Angew. Chem. Int. Ed., 2004, 43, 1546 61. M. Ko, Y. Kim, J. Woo, B. Lee, R. Mehrotra, P. Sharma, J. Kim, S. W. Hwang, H. Y. Jeong, H. Lim, S. H. Joo, J.-W. Jang, J. H. Kwak, Nat. Catal., 2022, 5, 37 62. L. Lu, B. Fang, Chem, 2022, 8, 1548 63. A. Zong, B. R. Nebgen, S. Lin, J. A. Spies, M. Zuerch, Nat. Rev. Mater., 2023, 8, 224 64. M. Dan, R. Zhong, S. Hu, H. Wu, Y. Zhou, Z. Liu, Chem Catal., 2022, 2, 1919 65. S. Kala Thirumalaikumaran, R. Suwathy, M. Venkatesan, J. Aerosp. Technol. Manag., 2018, 10, e2818 66. S. Corby, R. Rao, L. Steier, J. R. Durrant, Nat. Rev. Mater., 2021, 6, 1136 67. M. Y. Qi, M. Conte, M. Anpo, Z. R. Tang, Y. J. Xu, Chem. Rev., 2021, 121, 13051 68. Y. H. Li, Z. R. Tang, Y. J. Xu, Chinese J. Catal., 2022, 43, 708 69. Y. L. Wu, M. Y. Qi, C. L. Tan, Z. R. Tang, Y. J. Xu, Chinese J. Catal., 2022, 43, 1851 70. J. Y. Li, M. Y. Qi, Y. J. Xu, Chinese J. Catal., 2022, 43, 1084 71. Z. Zhang, T. Tsuchimochi, T. Ina, Y. Kumabe, S. Muto, K. Ohara, H. Yamada, S. L. Ten-no, T. Tachikawa, Nat. Commun., 2022, 13, 1499 -
Rights and permissions
This is an open access article under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Figures(7)
Information
Article Metrics

Export File
Citation
Xiaoming Xu, Yuanming Zhang, Yong Chen, Changhao Liu, Yang Li, Zhonghua Li, Zhetong Yang, Zhaosheng Li, Zhigang Zou. Green organic conversion with H2O2: challenges and opportunities[J]. Energy Lab, 2024, 2(1): 230011. doi: 10.54227/elab.20230011
Format
Content
 DownLoad:
DownLoad:
-
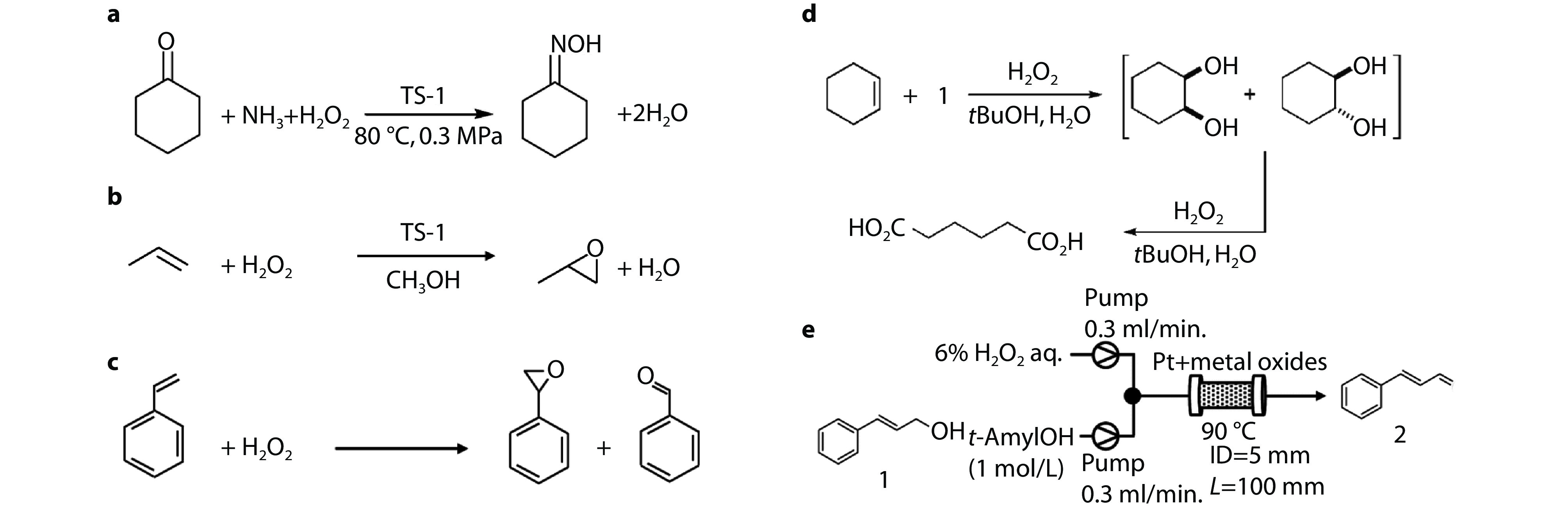
Figure 1.
a Reactions involved in cyclohexanone ammoximation methods.[5] Copyright 2021, Elsevier. b Industrial processes for propylene oxide production through the hydrogen peroxide propylene oxide (HPPO) process. c A plausible reaction pathway for the epoxidation of styrene. d Production of adipic acid with H2O2.[23] Copyright 2006, John Wiley and Sons. e Selective oxidation of (E)-cinnamyl alcohol with H2O2 using continuous flow reactors.[28] Copyright 2021, Royal Society of Chemistry.
-
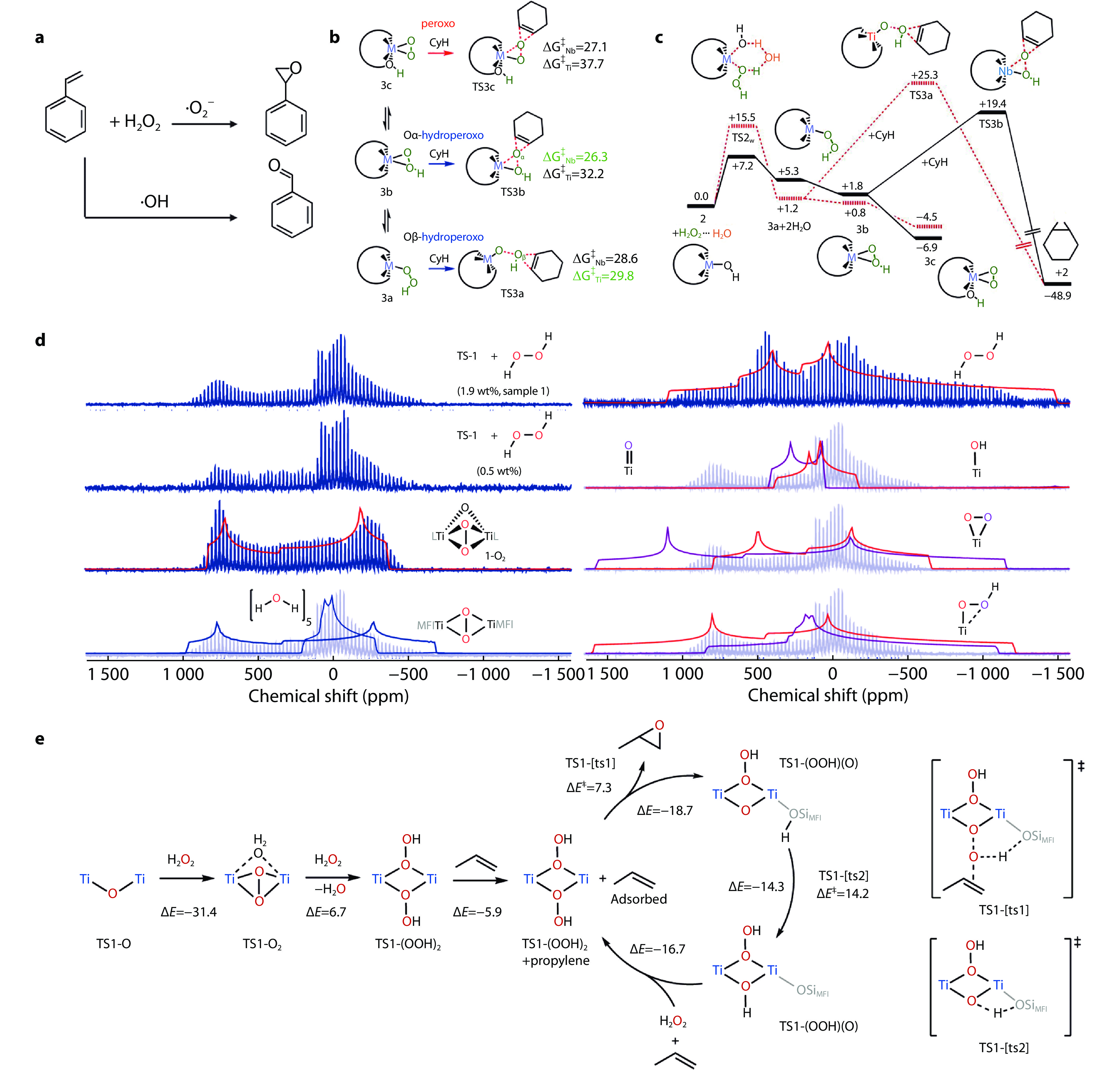
Figure 2.
a Reaction paths of styrene under different oxygen radicals. b Possible pathways for oxygen transfer to CyH from peroxo and hydroperoxo species in transition-metal-substituted polyoxometalates. c Calculated potential free-energy profile (kcal mol–1) for cyclohexene epoxidation with H2O2.[32] Copyright 2019, American Chemical Society. d Experimental and calculated solid-state 17O NMR spectra. e Calculated energy surface of propylene epoxidation at a dinuclear Ti site (electronic energies are given in kcal mol−1).[7] Copyright 2020, Springer Nature.
-
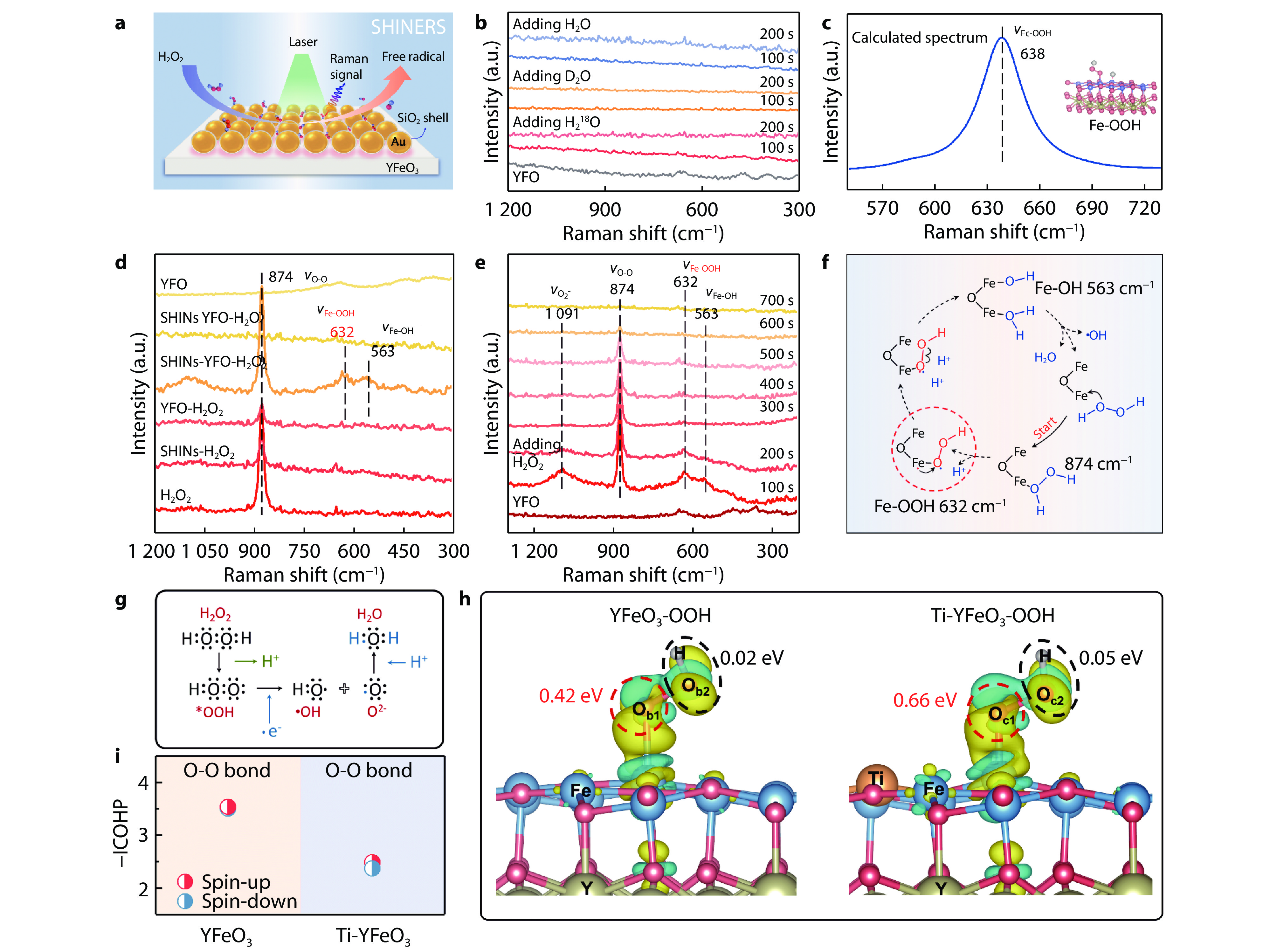
Figure 3.
a Schematic of shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) detection process. b SHINERS spectra of isotope labelling. c The correlated Raman frequencies of the Fe-O stretching vibration for Fe-OOH species (grey: H, red: O, blue: Fe and yellow: Y). d SHINERS spectra in different systems. e SHINERS spectra in the decomposition process of H2O2 as a function of time. f The possible mechanism for the activation process of H2O2. g Electron transfer process in the formation of •OH by H2O2 activation. h The different charge densities of *OOH adsorbed on YFeO3 (left) and Ti-YFeO3 (right). i The corresponding integral crystal orbital Hamilton populations (ICOHP) in YFeO3-OOH and Ti-YFeO3-OOH.[9] Copyright 2022, National Academy of Science.
-
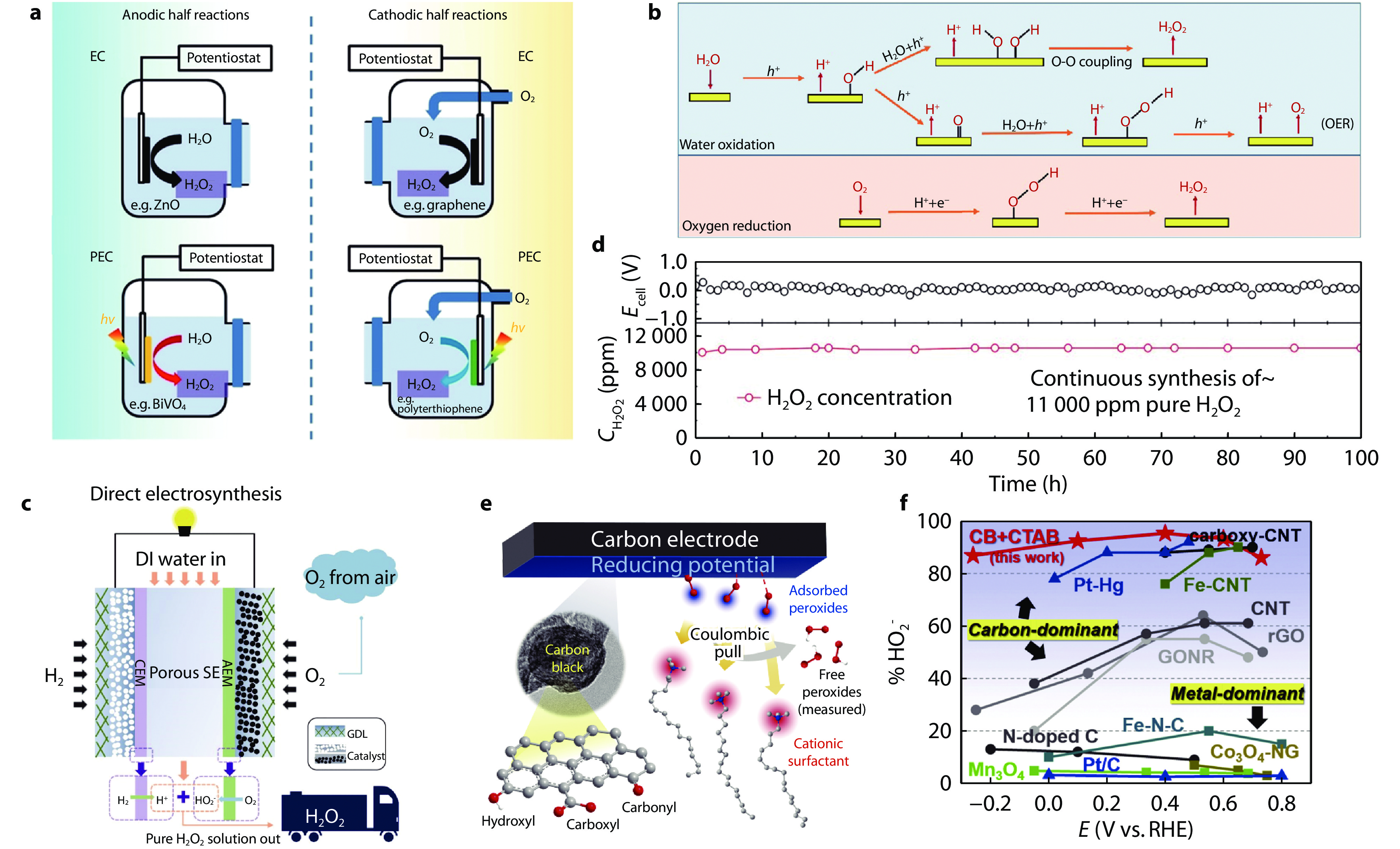
Figure 4.
a Schematic illustration of H2O2 production in a half-reaction achieved by water oxidation and oxygen reduction.[38] Copyright 2020, John Wiley and Sons. b Schematic illustration of ORR and WOR pathways.[40] Copyright 2022, Elsevier. c Electrochemical of H2O2 production using pure H2 and O2 streams separately introduced to the anode and cathode, respectively. d Stability tests for continuous generation of pure H2O2 solutions with concentrations >10,000 ppm. [44] Copyright 2019, American Association for the Advancement of Science. e In situ selectivity modulation by surface-acting cations at the carbon surface. f Comparisons of peroxide selectivity and potential window width among reported electrocatalyst systems.[45] Copyright 2020, Elsevier.
-
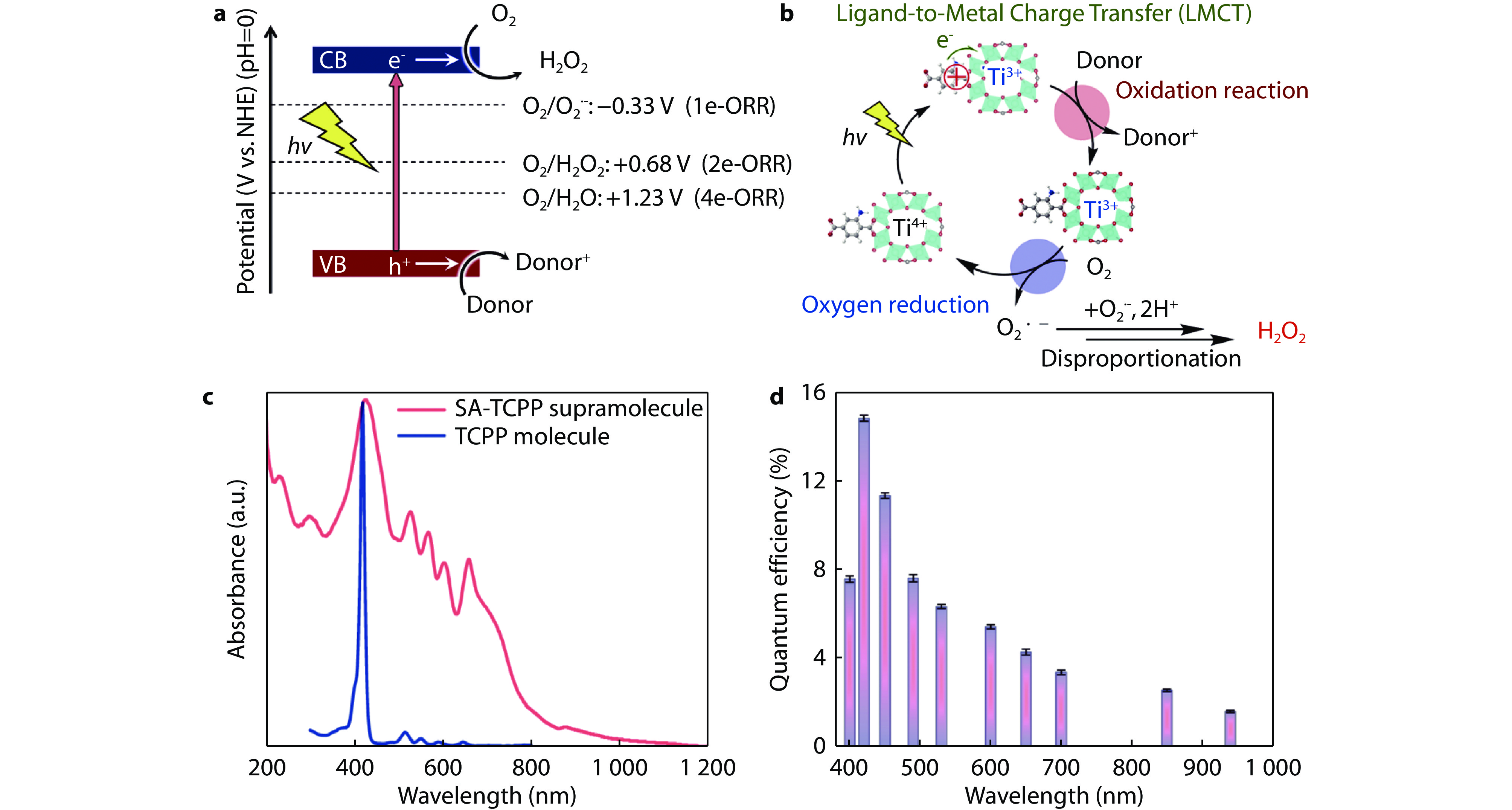
Figure 5.
a Energy diagram for photocatalytic H2O2 production via oxygen reduction. b Proposed reaction mechanism for photocatalytic H2O2 production.[47] Copyright 2022, Elsevier. c UV-vis-NIR DRS of SA-TCPP supramolecular assembly and UV–vis absorption spectrum of TCPP molecules. The strongest peak of SA-TCPP and TCPP molecules is located approximately at 415-430 nm. In contrast to TCPP molecules, the energy band absorption of SA-TCPP nanosheets extends up to 1,100 nm. d Quantum efficiency on SA-TCPP supramolecular photocatalysts with different bandpass filters.[48] Copyright 2023, Springer Nature.
-
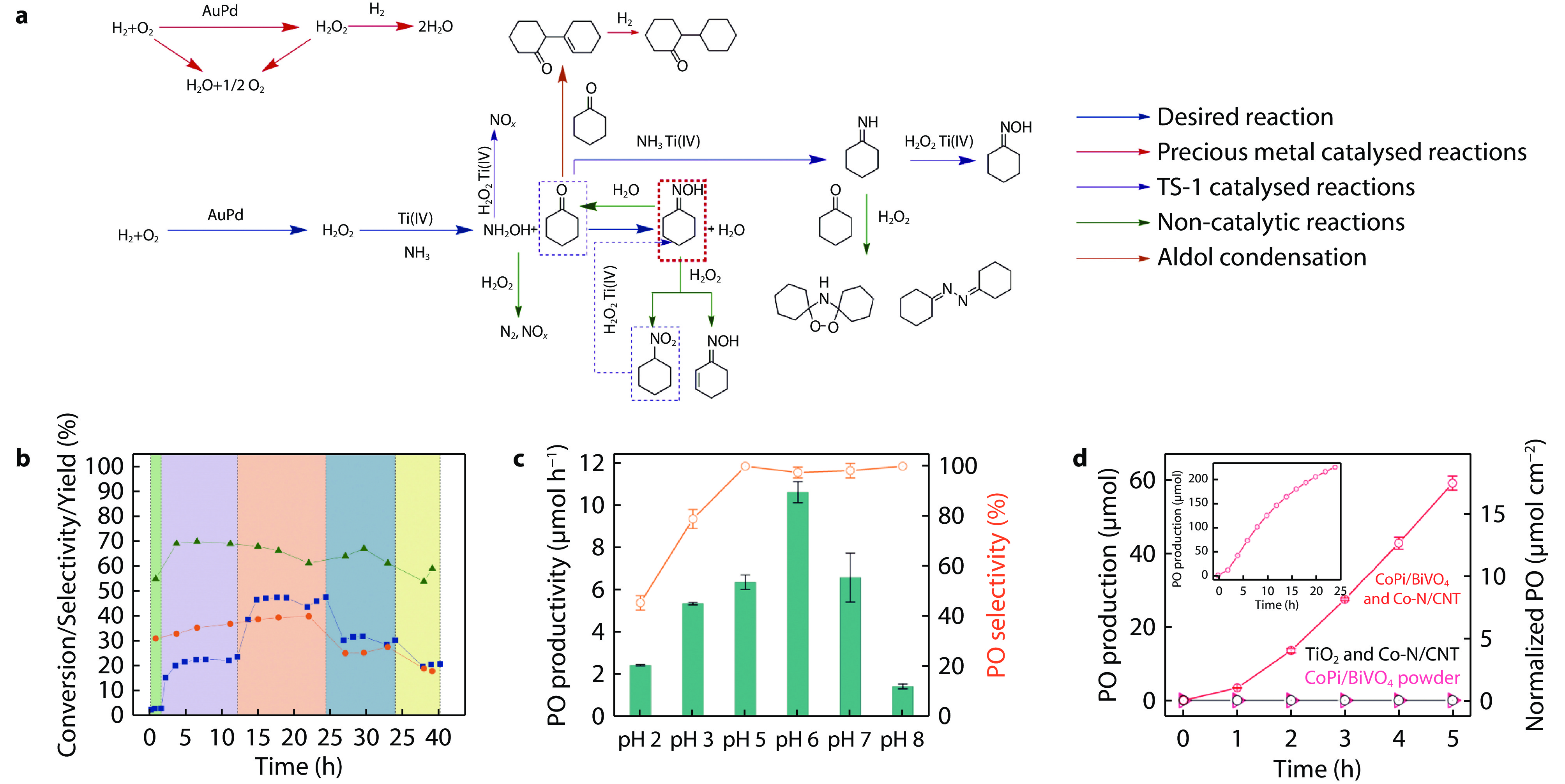
Figure 6.
a Proposed reaction pathways in the ammoximation of cyclohexanone-to-cyclohexanone oxime via in-situ-generated H2O2. In-situ H2O2 was generated on AuPd nanoparticles by binding with commercial TS-1 catalyst. And direct synthesis of H2O2 is combined with cyclohexanone ammoximation to produce oxime yields comparable to commercial processes. b Optimization of reaction parameters for the 0.33%Au-0.33%Pd/TS-1(Acetate-O+R) catalyst in a continuous regime. Cyclohexanone oxime yield (blue squares), H2 conversion (orange circles), H2 selectivity (green triangles).[13] Copyright 2022, American Association for the Advancement of Science. c PO production rate and PO selectivity after 5 h of reaction in solutions with different pH values. d PO production in the presence of photo-electro-heterogeneous catalytic and photo-heterogeneous catalytic systems. Inset: PO production for 24 h.[61] Copyright 2021, Springer Nature.
